Amiloidosi ATTR ereditaria: una malattia multisistemica, letale1-4
Gli effetti della malattia colpiscono diversi organi, causando vari sintomi1,3,4
Poiché le fibrille amiloidi si depositano nei tessuti di tutto il corpo, inclusi nervi, cuore e tratto gastrointestinale, i pazienti con amiloidosi ATTR possono presentare uno spettro di sintomi, sensitivo-motori, autonomici e cardiaci.1-5
Infatti, più della metà dei pazienti con amilodisi hATTR presentano un fenotipo misto.6,7
Variabilità di possibili segni e sintomi dell’amiloidosi hATTR
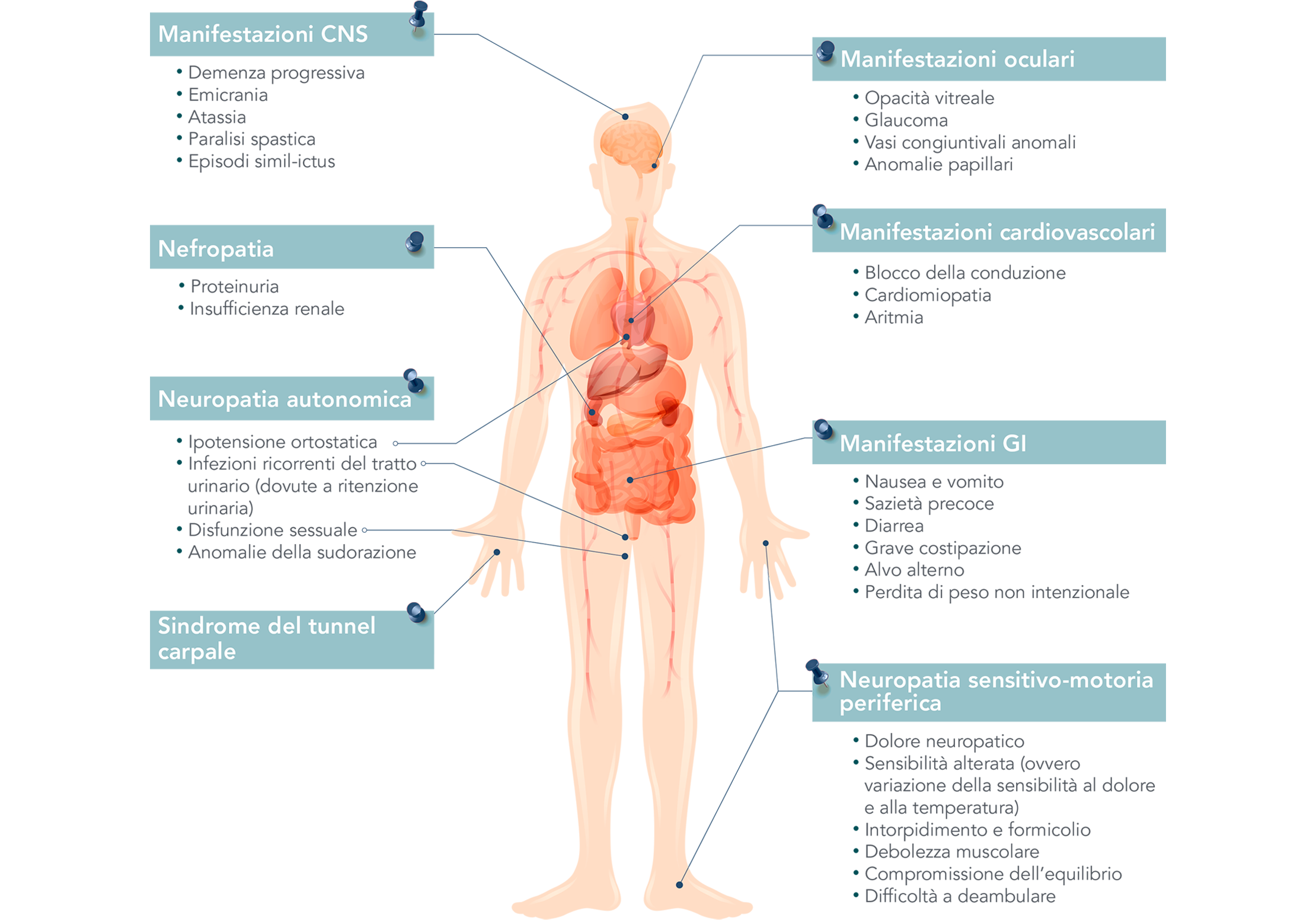
Adattato da Conceição I, et al. J Peripher Nerv Syst. 2016;21(1):5-9.
L’insorgenza dei sintomi è estremamente varia tra i pazienti, perfino all’interno della stessa mutazione. I sintomi inoltre possono variare tra pazienti della stessa famiglia8
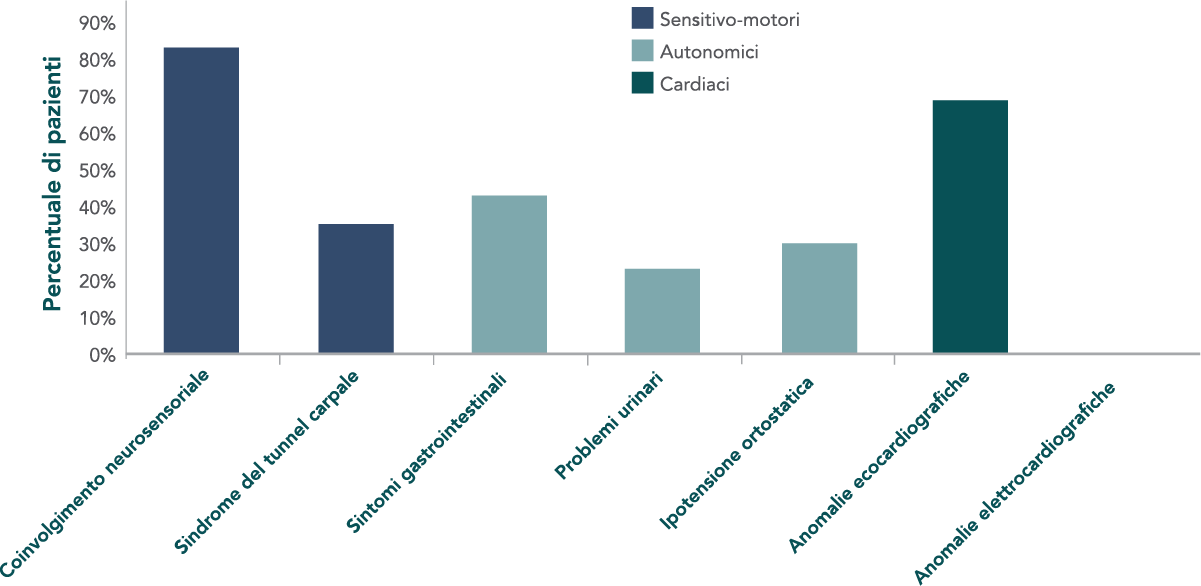
Caratteristiche cliniche di base di 186 soggetti con amiloidosi ATTR ereditaria in uno studio multicentrico.6