Amiloidosi ATTR ereditaria: una malattia familiare progressiva e potenzialmente letale1-3
Che cos’è l’amiloidosi hATTR?
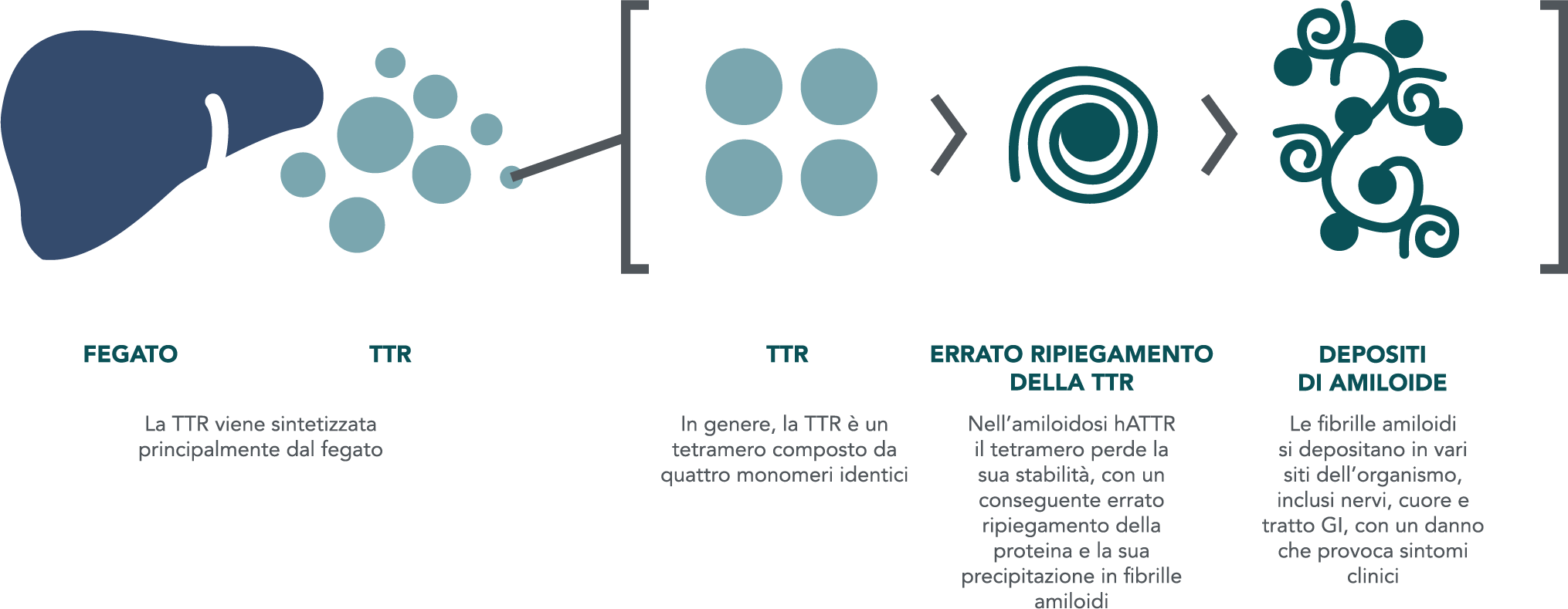
L’amiloidosi hATTR è una malattia autosomica dominante causata da una mutazione del gene transtiretina (TTR) che causa un accumulo delle proteine TTR mal configurate come fibrille amiloidi in più organi e sistemi inclusi i nervi, il cuore e il tratto gastrointestinale.2,4
Formazione delle fibrille amiloidi2,4,5
Bibliografia:
- Adams D, Coelho T, Obici L, et al. Rapid progression of familial amyloidotic polyneuropathy: a multinational natural history study. Neurology. 2015;85(8):675-682.
- Hanna M. Novel drugs targeting transthyretin amyloidosis. Curr Heart Fail Rep. 2014;11(1):50-57.
- Mohty D, Damy T, Cosnay P, et al. Cardiac amyloidosis: updates in diagnosis and management. Arch Cardiovasc Dis. 2013;106(10):528-540.
- Hawkins PN, Ando Y, Dispenzeri A, et al. Evolving landscape in the management of transthyretin amyloidosis. Ann Med. 2015;47(8):625-638.
- Sekijima Y. Transthyretin (ATTR) amyloidosis: clinical spectrum, molecular pathogenesis and disease-modifying treatments. J Neurol Neurosurg Psychiatry. 2015;86(9):1036-1043.
- Ando Y, Coelho T, Berk JL, et al. Guidelines of transthyretin-related hereditary amyloidosis for clinicians. Orphanet J Rare Dis. 2013;8:31.
- Rapezzi C, Quarta CC, Obici L, et al. Disease profile and differential diagnosis of hereditary transthyretin-related amyloidosis with exclusively cardiac phenotype: an Italian perspective. Eur Heart J. 2013;34(7):520-528.
- Adams D, Gonzalez-Duarte A, O’Riordan W, et al., an investigational RNAi therapeutic for the treatment of hereditary ATTR amyloidosis with polyneuropathy: baseline demographics from the phase 3 APOLLO study. In: The XVth International Symposium on Amyloidosis. Uppsala, Svezia: ISA International Society of Amyloidosis; July 3-7, 2016. PA 82.
- Coelho T, Maurer MS, Suhr OB. THAOS—The Transthyretin Amyloidosis Outcomes Survey: initial report on clinical manifestations in patients with hereditary and wild-type transthyretin amyloidosis. Curr Med Res Opin. 2013;29(1):63-76.
- Castaño A, Drachman BM, Judge D, et al. Natural history and therapy of TTR-cardiac amyloidosis: emerging disease-modifying therapies from organ transplantation to stabilizer and silencer drugs. Heart Fail Rev. 2015;20(2):163-178.